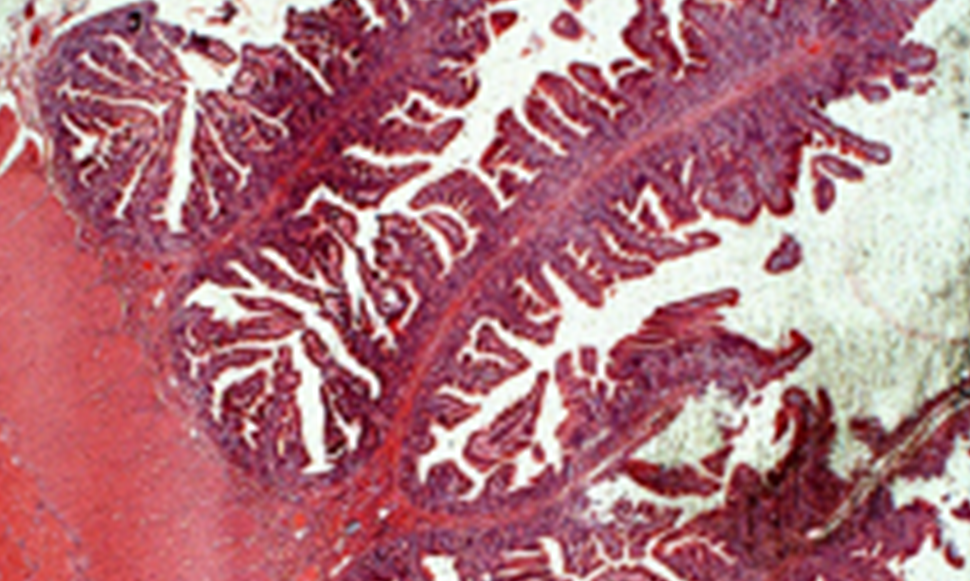
Invaginación intestinal secundaria a un tumor de GIST en una niña
DOI:
https://doi.org/10.31644/IMASD.43.2026.a08Palabras clave:
tumor de GIST en niños, causas de invaginación secundaria, sangrado de tubo digestivoResumen
Los tumores gastrointestinales estromales (GIST), son los tumores mesenquimales del tracto gastrointestinal más frecuente en adultos, pero son muy raros en niños. Nosotros reportamos el caso de una paciente femenina de 9 años con un cuadro de dolor abdominal y que el ultrasonido sugirió invaginación intestinal. Durante la laparotomía exploradora se encontró un tumor intraluminal en intestino delgado, que posteriormente por anatomía patológica confirmó el diagnóstico de tumor de GIST. No recibió tratamiento con quimioterapia. Se ha mantenido en observación por más de 5 años sin evidencia de metástasis o recidiva. Nosotros realizamos una breve discusión y análisis de la literatura del tumor de GIST en niños.
Descargas
Citas
Naito, Y., Nishida, T., & Doi, T. (2023). Current status of and future prospects for the treatment of unresectable or metastatic gastrointestinal stromal tumours. Gastric Cancer, 26(3), 339-351. https://doi.org/10.1007/s10120-023-01381-6
2.- Tyagi, I. V., & Anand, S. (2025). Gastrointestinal Stromal Tumors Market Share, Report 2035. En Market Research Future. https://www.marketresearchfuture.com/reports/gastrointestinal-stromal-tumors-market-1583/?utm_term=&utm_campaign=&utm_source=adwords&utm_medium=ppc&hsa_acc=2893753364&hsa_cam=23142125492&hsa_grp=190076755354&hsa_ad=779362054048&hsa_src=g&hsa_tgt=dsa-2443880216606&hsa_kw=&hsa_mt=&hsa_net=adwords&hsa_ver=3&gad_source=1
3.- Popoiu, T., Pîrvu, C., Popoiu, C., Iacob, E. R., Talpai, T., Voinea, A., Albu, R., Tãban, S., Bãlãnoiu, L., & Pantea, S. (2024). Gastrointestinal Stromal Tumors (GISTs) in Pediatric Patients: A Case Report and Literature Review. Children, 11(9), 1040. https://doi.org/10.3390/children11091040
4.- Hallie J. Quiróz, Brent A. Willobee, Matthew S. Sussman, Bradley R. Fox, Chad M. Thorson, Juan E. Sola et al. Pediatric gastrointestinal stromal tumors- a review of diagnostics modalities. Transl Gastroenterol Hepatol. 2018;3:3-54 doi:10.21037/tgh.2018.07.08
5.- M. Shimomura, S. Ikeda, Y. Takakura, Y. Kawaguchi, M. Tokunaga, Haruka Takeda, et al. Gastrointestinal stromal tumors of the small intestine in pediatric populations: a case report and literature review. Pediatr Surg Int, 26 (2010), pp. 649-654
6.- Kinblom L-G, Remotti HE, Aldenborg F, et al. Gastrointestinal Pacemeaker cell tumor (GIPACT). Gastrointestinal stromal tumors show phenotypic characteristics of interstitial cells of Cajal. Am J Pathol 1998; 152: 1259-69. 5.
7.- Saund MS, Demetri GD, Ashley SW. Gastrointestinal stromal tumors (GISTs). Curr Opin Gastroenterol 2004; 20: 89-94.
8.- Corless CL, Fletcher JA and Heinrich MC. Biology of gastrointestinal stromal tumors. J Clin Oncol 2004; 22: 3813-25
9.- Bucher P, Villiger P, Egger J-F, et al. Management of gastrointestinal stromal tumors: from diagnosis to treatment. Swiss Med Wkly 2004; 134: 145-53
10.- Miettinem M, Lasota J. Gastrointestinal stromal tumors (GISTs): definition, ocurrence, pathology, differential diagnosis, and molecular genetics. Pol J Pathol 2003; 54: 3-24.
11.- Inga-Marie Schaefer, Adrián Mariño Enríquez, Jonathan A Fletcher. What is new in gastrointestinal stromal tumor?. Adv Anat Pathol. 2017;24(5): 259-267
12.- Morales Peralta Adrián, Covarrubias Espinoza Gilberto, Rios García Candy Guadalupe, Larios Farak Tania Clarisa, Millán Valenzuela Luis Omar, Galván Ruiz Vanessa Guadalupe. Tumor de GIST pediátrico. Presentación de dos casos y revisión de la literatura. Bol Clin Hosp Infan Edo Son. 2017;34(2): 127-135
13.- B. Samarji, T. Walter, F. Dijoud, S. Collardeau Frachon, F. Hameury, R. Dubois, et al. [Pediatric gastrointestinal stromal tumors: report of three cases] Gastroenterol Clin Biol, 34 (2010), pp. 407-40
14.- Stiles, Z.E.; Fleming, A.M.; Dickson, P.V.; Tsao, M.; Glazer, E.S.; Shibata, D.; Deneve, J.L. Lymph Node Metastases in Gastrointestinal Stromal Tumors: An Uncommon Event. Ann. Surg. Oncol. 2022, 29, 8641–8648.
15.- Ng EH, Pollock RE, Munsell MF, Atkinson EN, Romsdahl MM. Prognostic factors influencing survival in gastrointestinal leiomisarcoma. Implications for surgical management and stag ing. Ann Surg. 1992; 215: 68-77
16.- Prof Yoon-Koo Kang, Min-Hee Ryu, Changhoon Yoo, Prof Baek-Yeol Ryoo, Hyun Jin Kim, Jong Jin Lee, et al. Resumption of imatinib dosing to control metastatic gastrointestinal Stromal Tumors (GIST) after failure of Imatinib and Sunitinib: Results of a randomised, placebo-controlled, phase 3 trial (RIGHT). Lancet Oncol. 2013; 14(12): 1175-1182. doi:10.1016/S1470-2045(13)70453-4.
17.- Arimatias Raitio, Adeline Salim, Dhanya Mullassery, Paul D Losty. Current treatment and outcomes of pediatric gastrointestinal stromal tumors (GIST): a systematic review of published studies. Pediatr Surg Int. 2021; 37(9):1161-1165
Publicado
Versiones
- 18-03-2026 (2)
- 03-02-2026 (1)
Cómo citar
Número
Sección
Licencia
Derechos de autor 2026 Universidad Autónoma de Chiapas

Esta obra está bajo una licencia internacional Creative Commons Atribución-NoComercial-CompartirIgual 4.0.